New work on Tree/dendrogram
view
It was
found that the Tree View software by Eisen et al (1998) suits well to
situations requiring very accurate clustering like the phylogenetic
classification. However, three major changes were sought to the basic
algorithm, viz., the generation of tree for clustering more than two similarly
expressing genes, reducing the accuracy of the gene expression matrix and
taking input from the gene expression matrix instead of the distance matrix.
In this context, the Tree View module was re-written to incorporate these
changes, without jeopardizing on the accuracy of visualization. The
output of the modified/revised version integrated into the GEDAS software was
comparable to the Tree View software by Eisen et al. Following were the
observations:
· The Tree View software was generating a binary tree representation for all nodes (or genes), hence making it a perfect dendrogram. However, it is quite but natural that the entities (genes in this case) in reality exhibit similarity and belongingness to a particular group (or cluster). In short, the genes exhibit behaviour similar to human society. Just as there can be more than two children in the same family, the concept does apply to genes as well. In such a situation, the prevailing tree or dendrogram does not suit well to the clustering of gene expression data.
Representation of more than two genes falling under the same cluster head would mean that their behaviour (or function) as well as expression level has been identical. In order to bring more and more genes under the same grouping should be the motive behind the clustering and visualization. The only exception where the old Tree View software applies suitably is for clustering of genes based on a negligible change in one or more nucleotides of genes or motifs of proteins. For instance, when genes of human across the world are compared, the fair grouping generated would relate to races such as Asian, African, European, etc. and not as Indian, Bhutanese, Bangladeshis, Sri Lankan, etc. since persons of these nations have identical genes. Therefore, branching of genes (be it gene expression data) within any given cluster further down till the last two genes would be absurd.
· It was also observed that the Tree View software was very sensitive even to a fractional change in the distance matrix, which was not essential for clustering. Clustering, as the term defines, means general grouping of genes together; and it is possible to obtain such grouping even with lesser precision of the data.
A step ahead, the difference between gene expression values of 1.2 and 1.3 does not either provide significant information about the gene expression nor useful in properly clustering the genes. This fractional change of 0.1 is too small to be considered in depth when analysis of thousands of genes and grouping into handful of clusters has been considered. The dendrogram (Tree View) thus generated has been confusing, and not giving substantial information on clusters and sub-clusters.
· It was further observed that the Tree View software was generating the dendrogram (both the tree part and the checks view part) using the distance matrix only, which it was felt was an inaccurate representation and the checks view (sometimes also called the heat map) part was generated using the preprocessed gene expression data matrix.
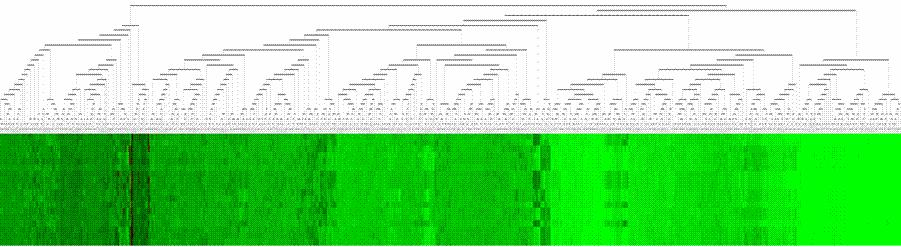
The above changes brought great change in the visualization over the old software. A sample of both has been produced below for the same dataset (code tree and gene tree files), as given in following figure:
|
|
|
|
|
|
|
|
|
Figure: Comparison of Tree View outputs for Arabidopsis thaliana dataset (log transformed) using HC (complete linkage) (a) Eisen Lab’s Tree View output (b) Revised/new Tree View output. Note the clarity in cluster output, leaving aside the colour coding scheme, both the tree and the checks strip have been enhanced. The graphical representation of distances in the tree do not play vital role in microarray informatics as the purpose is not of bringing out phylogenetic comparison |